4HF Biotec runs programs to identify new targets for all solid and hematological cancers using our 4HF CancerDataMiner™ platform. In parallel, small molecules binding to our prioritized cancer-related targets are currently under identification using computer-aided drug design methodologies. The use of our proprietary platform, several in silico tools, and databases allows to optimally determine the best indications, drug performance and predictive biomarkers

Target Identification
We prioritize novel cancer targets that are most likely to be druggable with conjugate approaches. We favour drugs most likely to be effective against specific cancer types, and our approach anticipates potential side effects. Deeper analysis of the best candidates includes gene expression across each tumor entity, gene functions and regulatory mechanisms, providing valuable insights into the underlying complex biology of cancers.
Because the success of these drug-conjugates depends heavily on the presence of the target protein on the cancer cell surface, candidates are investigated at the protein level, taking into account parameters such as expression levels, localization, stability, degradation pathways, and recycling characteristics.
Finally, the questions of the protein structure and its druggability are addressed to prepare next step of research to identify ligands that will form our conjugates.
Small Molecule Identification
4HF leverages state-of-the art computational methods for the identification of small molecules binding selectively the cancer targets of interest. Advances in hardware as well as in AI-driven protein structure prediction and virtual drug screening have rendered reality the accurate screening of millions of compounds in a matter of weeks. We are committed to push the limits of what’s possible in drug discovery, by continuously expanding our directed screening approach. While conventional in silico drug screening campaigns start right away with virtually screening a target with a broad dataset of small molecules, our approach leans on the idea that we want to first clearly define a chemically optimal binding pocket and then create a focused small molecule library to screen against that specific binding pocket.
The overall process by which 4HF screens for new small molecule drug candidates can be separated into three steps:
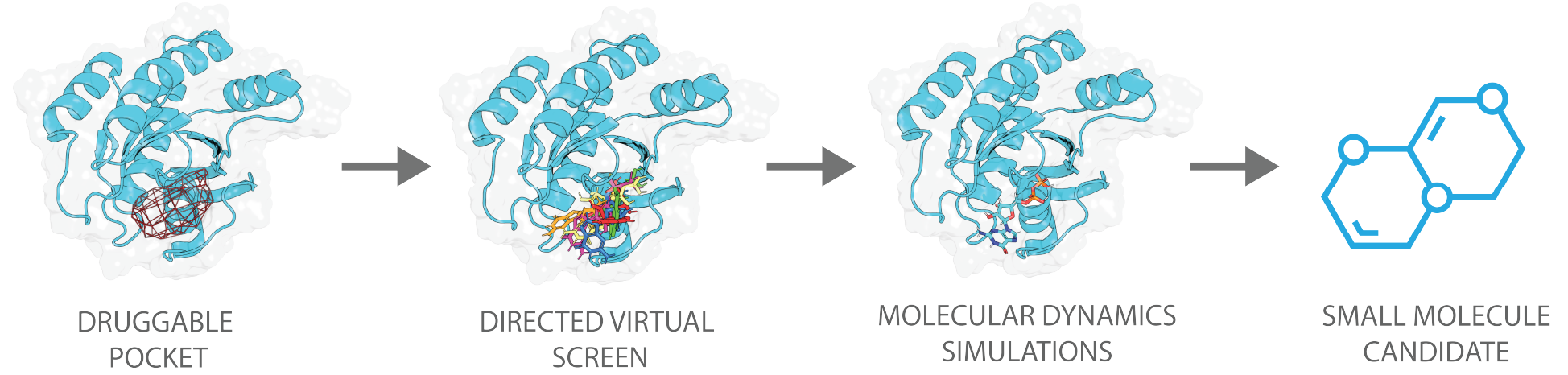
In the first step, a thorough structural analysis is carried out on the target incorporating cutting-edge AI models and advanced molecular dynamics simulation protocols to find the optimal binding pocket. With this, a pharmacophore* hypothesis is generated, which is then used to generate a focused library of small molecules that is tailored to the selected binding pocket of the target. Subsequent virtual screening hits are ranked and analyzed for their potential as a drug. In the last step, the most promising hits are then further validated through an in-depth analysis with molecular dynamics simulations.
Our selected small molecule candidates will then enter in the experimental phases to confirm binding and to identify the ligands that strongly bind the cancer-associated protein target.
* Properties that are necessary for a ligand to be noncovalently bound by a protein
Drug Conjugates Development
Following target and ligand validations, the confirmed ligands are conjugated with the optimal combination of linkers and cytotoxic payloads.
The resulting Small Molecule Drug Conjugates (SMDCs) are then profiled for target binding in target-expressing tumor cells, internalization into lysosomes, cytotoxic effects on tumor cell lines in culture, stability in plasma, pharmacokinetics and tissue distribution in mice and finally efficacy and tolerability in tumor-bearing mice. The goal is, in each case, to identify a clinical candidate. At that point at the latest, we look for partners for clinical development.
Depending on the target, we are also open to drug conjugate formats other than SMDCs, such as Antibody Drug Conjugates (ADCs), radiopharmaceuticals, bispecific antibodies or CAR-T cells.